


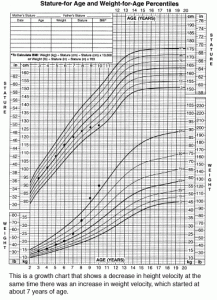
Figure A
How is Cushing’s syndrome (CS) in children different than in adults?
- CS in children is rare. An estimated 10 to 15 of every million people are affected a year and only about ten percent of these new cases occur in children.
- For pre-pubertal children, and certainly for toddlers, adrenal tumors (e.g. adenoma, carcinoma, or bilateral hyperplasia) are a more common cause of CS. In older children, pituitary adenomas are a more common cause of CS. Ectopic (a site other than pituitary or adrenal) sources of CS are extremely rare in children. Overall, there is a female to male predominance, which decreases with younger age.
- There is no single symptom shared by every child with CS. It is common for children with CS to show an increase in rate of weight gain accompanied by a decrease in growth rate (linear height). Figure A shows a growth chart that is typically seen in children with CS. Other problems that may occur in children with CS include: rounded face, reddened cheeks, acne, headache, excess hair growth (fine downy hair on cheeks, arms, and legs), purplish-pink stretch marks (more common in older children), darkened skin around neck and armpit areas, easy bruising, development of pubic hair at a younger age than usual, irregular or absent menstrual periods, and high blood pressure. Figure B shows a photograph of a girl with CS standing next to her twin brother who does not have CS. Compared to adults with CS, symptoms that are less commonly seen in children include: sleep disruption, mental changes, and muscle weakness. Although many adults with CS report change in mental status that affect their job performance, children with CS do not usually report problems with school performance.

Figure B
What should I do if I think my child might have CS?
- Schedule an appointment with a pediatrician or pediatric endocrinologist immediately. Take pictures that show what your child looked like prior to the start of the symptoms of possible CS. As with adults with possible CS, the diagnosis is not always easy.
- The diagnosis of CS is based on a review of your child’s medical history (including possible exposure to medications or other substances that contain steroids), physical examination, review of growth data (growth chart), and laboratory tests. Ask your child’s doctor to review your child’s growth chart with you. If the growth chart shows an increase in the rate of weight gain and decrease in the rate of linear growth (height velocity) during the same time period, then further evaluation and monitoring is needed to determine the cause.
How is CS diagnosed?
It is important for children to be evaluated by a pediatric endocrinologist who is experienced with CS, since some of the tests require adjustment for the pediatric patient. When the diagnosis of CS is suspected, it is important to document hypercorticolism (high cortisol levels).
The best way to screen for elevated cortisol levels in children is a 24-hour urine collection for urinary free cortisol (UFC) determination. Frequently, more than one 24-hour urine collection is advised for screening. If the results of the 24-hour UFC show elevated values, then a referral to a pediatric endocrinologist is advisable. It is important to remember that in order to correctly interpret the results of the 24-hour UFC, the result must be “corrected” for the child’s body size ( or ‘body surface area’, a mathematical formula that is based on the child’s height and weight). High UFC values can be caused by other conditions, such as physical and emotional stress, obesity, pregnancy, chronic exercise, depression, etc. These conditions may cause what is known as pseudo-Cushing’s syndrome. Although this is more common in adults, it is frequently seen in children, especially in obese children or adolescents with other medical or psychological problems.
Salivary cortisol levels (collecting saliva in a tube at midnight and in the morning) are also used as a screening test. Normally, the cortisol level in the body is very low at midnight (when we are in the deepest of the sleep phases), and higher in the morning. With CS, the body loses the diurnal pattern (day vs. night) and the cortisol levels at midnight are higher than expected; often not very different from the early morning cortisol level.
Another screening test is the low-dose dexamethasone test, which involves giving one milligram of dexamethasone (adjusted for the child’s weight) and measuring the morning cortisol level in the blood. Results of a study done at N.I.H. show that in the morning, the cortisol level should be less than 5mcg/dL. Recent data in adults suggests a value less than 1.8mcg/dL rules out Cushing’s. The low-dose dexamethasone test has not been validated extensively in children.
Once the diagnosis of CS is confirmed, additional testing is needed to determine the cause. If possible, it is best to have the diagnostic evaluation done in a pediatric endocrine center experienced with the evaluation of CS. Testing is done to determine whether the CS is due to an adrenal, pituitary, or ectopic cause (ACTH dependent or not). It is beyond the scope of this article to review in detail the various diagnostic tests used in the evaluation of CS. Some tests are done to confirm the diagnosis of CS and some tests are done to distinguish the specific cause of CS. Please refer to the CSRF web site for more specific information about these tests.
Imaging studies are an important tool in the diagnostic evaluation of CS. When CD is suspected, magnetic resonance imaging (MRI) of the pituitary (with contrast enhancement) should be performed. Generally, pituitary MRI may detect only about half of ACTH producing pituitary tumors; however, recently, at the National Institutes of Health, a new MRI technique has been developed (SPGR-MRI) that may increase sensitivity to 70-80%. Finally, computed tomography (CT) of the adrenal glands is helpful in the evaluation of pituitary versus adrenal causes of CS.
Treatment
- Treatment for CS depends on the specific cause of the cortisol excess. Referral to a pediatric endocrinologist who is experienced with the diagnosis and treatment of CS is very important. Prolonged exposure of the body to excess cortisol levels has many negative effects, including: obesity, loss of final height, high blood pressure, osteoporosis and other orthopedic problems, diabetes, and increased susceptibility to infections.
- Pituitary tumors (adenomas) are usually treated by surgical removal, which is known as transsphenoidal surgery (TSS). Due to the highly specialized nature of this surgery, referral to a neurosurgeon who is experienced with this procedure is recommended. In certain patients, treatment with medication(s) that control or block cortisol production, may be done on a short-term basis. In some patients, when surgery has failed (or if they were not good candidates for surgery, which is very rare in children but more frequent in adults), radiation therapy is indicated.
- Adrenal tumors and bilateral hyperplasia (bilateral adrenal nodular disease, primary pigmented adrenal nodular disease) are treated surgically, by either unilateral or bilateral adrenalectomy, as appropriate.
Will my child lead a normal life after treatment for CS?
- Various studies report that one year after surgical cure of CS most children had lost weight and body mass and their height and growth velocity had increased; however final adult height is often impaired (by at least an inch or more).
- Risk factors associated with TSS for removal of a pituitary adenoma include temporary or permanent dysfunction of the pituitary gland. Therefore, it is important for the child and adolescent to be monitored on a routine basis by a pediatric endocrinologist to screen for any problem with pituitary gland function, including hypothyroidism, adrenal insufficiency, growth hormone insufficiency, pubertal delay. After TSS for CS, daily cortisol replacement is necessary, typically for a period of six to eighteen months, until the hypothalamic-pituitary-adrenal axis (HPA) recovers, Many children often experience some symptoms of steroid withdrawal during this period (e.g. fatigue, headache).
- Children and adolescents who undergo bilateral adrenalectomy for treatment of CS experience a recovery period that is similar to other patients who undergo major abdominal surgery (depending on whether the procedure is able to be performed by laproscopy or not). Because both adrenals are removed, these children will require daily hormone replacement (hydrocortisone and fludrocortisone) for the rest of their life. In addition, the hydrocortisone dose will require adjustment during illness episodes and significant physical stress (e.g. surgery, trauma).
- Children with an adrenal tumor that requires the removal of a single adrenal need to take daily hydrocortisone replacement until the HPA axis recovers, just like patients with surgically resected pituitary tumors.
- Most children and adolescents who are recovering from CS are able to resume normal physical activities within several weeks to months. Many children and adolescents recovering from CS experience changes in cognitive performance that can be stressful for both the child and the parents. The brain is affected by prolonged exposure to abnormally high cortisol levels and once the cortisol levels are normalized there is a period of readjustment. Symptoms reported by some children and adolescents include difficulty concentrating and problems with memory that may affect their academic performance for an indeterminate period. It is important to provide appropriate educational and psychological resources for the child or adolescent during this period.
Editor’s Note: Meg Keil, MS, CRNP is a nurse practitioner at the NIH in Bethesda, MD in the pediatric endocrinology division. She is an associate investigator for two protocols for the evaluation and treatment of pediatric Cushing’s at NIH. (Dr. Constantine Stratakis, a pediatric endocrinologist, is the principal investigator for these protocols.) She can be reached at: 301-435-3391 or email: [email protected].